de novo ゲノムシーケンス

新規ゲノム配列決定では、種についての最初のゲノムマップは系統発生の研究、種の多様性、特定の形質と遺伝子マーカー、および他のゲノミクス研究のマッピングを分析するための貴重な基準配列を提供し、生成されます。
次世代シーケンシング技術の発展に伴い、新規のゲノム配列決定は新規ゲノム配列上のいくつかの重要な出版物に貢献し提供することができます。
サービスの強み
- Highly experienced
- We have completed major de novogenome sequencing projects, and our data has been published in top-tier journals.
- Leader in NGS services
- We provide high-quality sequencing, an efficient standard workflow, fast turnaround time, and bioinformatics analyses at a cost-effective price.
- Bioinformatics expertise
- he SOAPdenovoII software and the NovoHeter software, developed by Novogene scientists, are used for complex genome assembly.
Project Types
- Simple Genome
- Simple genome refers to a haploid genome with a low repeat content (less than 50%), or a diploid genome with a low rate of heterozygosity (less than 0.5%), such as most mammals, birds, and cultivated crops
- Complex Genome
- Complex genome refers to a diploid or polyploid genome with a high repeat content (higher than 50%) or a high rate of heterozygosity (higher than 0.5%), such as many species of plants, aquatics, and insects.?Moderately heterozygous genome (diploid)?Highly heterozygous genome (diploid)?Highly repetitive genome (diploid)
サービスの流れ
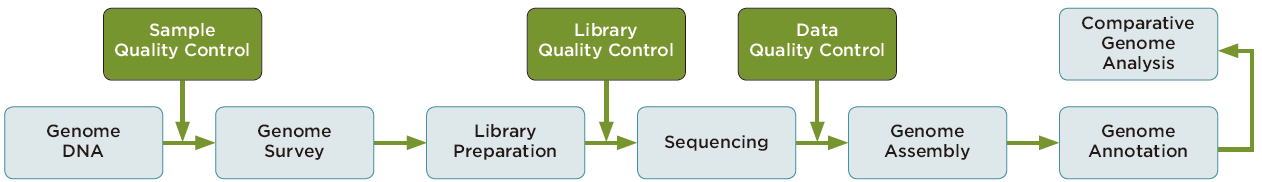
- SEQUENCING STRATEGY
- HiSeq platform, paired-end 150 bp
- SAMPLE REQUIREMENTS
- DNA amount for survey: ≧ 10 μg (quantified by Qubit 2.0)
- DNA amount for RAD-seq: ≧ 3 μg (quantified by Qubit 2.0)
- DNA amount for genome de novo sequencing: ≧ 500 μg (quantified by Qubit 2.0
- DNA concentration: ≧ 50 ng/μ
- Purity: OD260/280 = 1.8 - 2.0 without degradation and RNA contamination
- DATA QUALITY GUARANTEE
- Simple genome: Contig N50 ≧ 30 Kb, Scaffold N50 ≧ 1 Mb
- Mammal or bird genome: Contig N50 ≧ 30 Kb, Scaffold N50 ≧ 3 Mb
- Complex genome: Contig N50 ? 20 Kb, Scaffold N50 ? 300 Kb
- ヒトゲノム
- 全ゲノムシーケンス
- 全エクソームシーケンス
- 遺伝子制御
- RNA-seq(mRNA / LncRNA)
- メチル化DNA解析
- ChIP-seq
- 動植物ゲノム
- de novoシーケンス
- リシーケンス